
苏州纳米所蔺洪振团队Adv. Sci.:镁硫电池中串联催化研究取得进展
发布日期:2025-06-05 浏览次数: 【大中小】 【关闭】
二次电池因其能量转换效率高、使用寿命长和潜在的低成本而在储能领域具有广阔前景。在过去的十年中,已经研究了各种新型电池,包括碱金属-硫电池,例如室温(RT)钠-硫(Na-S)和钾-硫(K-S)电池,以及其他金属-硫电池,例如镁-硫(Mg-S)、铝-硫(Al-S)、锌-硫(Zn-S)和钙-硫(Ca-S)电池。然而,新兴的金属硫电池面临着严峻而复杂的挑战,与锂离子电池(LIB)相比,镁离子电池(MIB)其输出的体积容量比LIB高两倍。同时,镁在地球上资源丰富,环境友好。然而,镁电池的开发仍然存在一些关键问题,严重阻碍了可充电镁电池(RMBs)的商业化。目前阻碍Mg-S电池实际应用的障碍主要体现在:不导电的硫正极反应动力学缓慢、相容性电解质的设计、镁负极抗钝化的表面优化等。众所周知,Mg2+与电解质中的溶剂配位形成大的溶剂化结构,并且需要在Mg2+参与电化学反应之前或期间脱溶剂。在电极/电解质界面上,脱溶剂步骤和正极或负极氧化还原步骤相互耦合,可视为不同途径的级联反应。然而,目前对Mg-S电池正极的研究现状及进一步发展仍然缺乏系统而深入的总结和分析。
结合前期镁/锂/锌电池的相关研究基础(Energy Environ. Sci.,2024,17,3765-3775; Adv. Funct. Mater.,2025,2506397; Adv. Mater. 2023,35,2302828;Adv. Funct. Mater. 2023,2302624;ACS Nano 2023,17,2,1653;Adv. Funct. Mater. 2023,2212499; InfoMat. 2024,6,e12558; Adv. Mater. 2025,2501079;Nano Lett. 2025,25,3756-3765;Adv. Mater. 2024,2402792;Adv. Sci. 2024,2401629),中国科学院苏州纳米所蔺洪振研究员联合德国卡尔斯鲁厄理工学院王健博士综述了Mg2+脱溶、Mg2+在界面和正极内部的迁移以及硫转化等高能垒过程的研究进展,突出了上述过程之间可能的耦合方式,进一步综述了在提高电导率的前提下加速脱溶剂和硫转化动力学的级联催化策略。
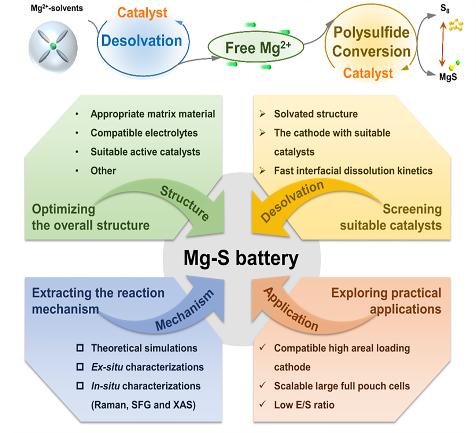
由于二价离子的高电荷密度, Mg2+与溶剂会形成大的溶剂化结构,并产生具有巨大空间效应的团簇,不仅阻碍了Mg2+在电解液中的迁移,而且导致反应动力学连续缓慢。在转化型电极上进行氧化还原转化反应的一个必要条件是溶剂化Mg(solvents)x2+的快速解离,虽然镁离子的脱溶究竟发生在反应的哪个阶段还不清楚,但反应的起始和终止状态是一致的,也是确定的,即从固态硫到液态多硫化物,最后转化为固态MgS,那么无论中间过程的载体形式如何,脱溶必然发生在整个反应过程中,因此去溶剂化是不可避免的。
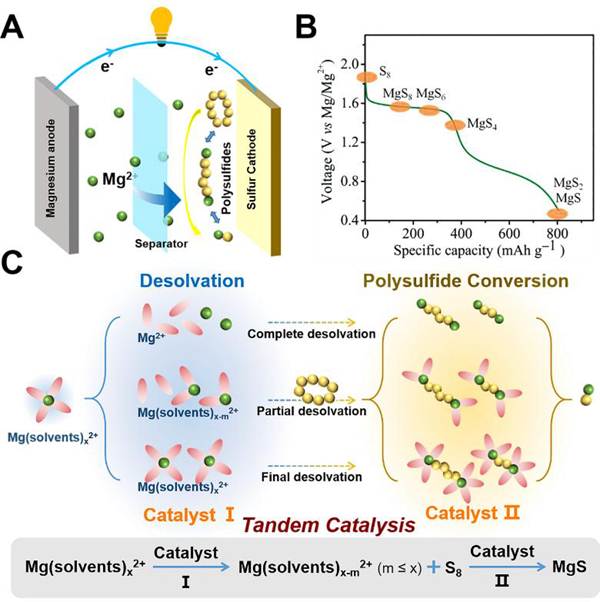
图1. Mg-S电池放电过程中硫正极上最常见的反应途径及可能的去溶剂化过程和典型的串联催化反应的示意图
在实际电池环境中,在到达硫正极之前,Mg2+倾向于与电解质中的溶剂分子合作以形成Mg(solvents)x2+。在硫/电解质界面处,大尺寸的Mg(solvents)x2+团簇首先解离形成Mg2+,这是多硫化物氧化还原转化反应的前提。因此,Mg2+在界面或正极内部的去溶剂化和扩散行为与Mg(solvents)x2+溶剂化结构的状态密切相关。将配位溶剂分子从Mg(solvents)x2+溶剂化鞘中解离出来对于形成自由分离的Mg2+显得非常重要。一般来说,从Mg(solvents)x2+中直接分离溶剂分子涉及相关化学配位键的断裂,从而呈现高能量势垒。因此,有必要降低用于形成Mg2+的界面屏障以平滑地扩散穿过正极/电解质界面。
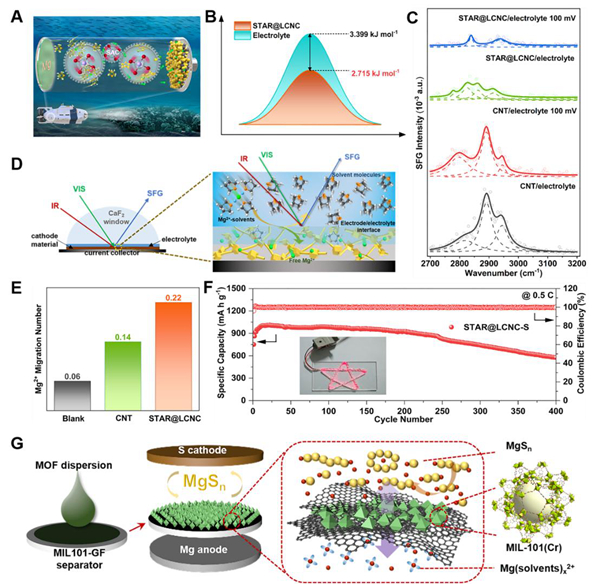
图2. 催化剂促进Mg2+的去溶剂化和扩散的动力学
Mg-S化学反应是发生在导电基质和电解质之间的界面处的大多数界面反应。为了提高硫的利用率,应扩大导电碳基体/电解质/硫三相界面的重叠区域,并在三相界面引入合适的催化位,以促进Mg2+与S的结合,促进后续的硫/硫化物转化。同时需要注意的是,应设计合适的碳结构或催化位点,限制多硫化物的扩散,防止它们穿梭到镁负极侧形成“死硫”。还需要确保催化剂充分暴露而不被硫化物覆盖和钝化,以有效增强Mg-S电池的可逆性。双电层(EDL)由扩散层和亥姆霍兹层组成,其中亥姆霍兹层又分为内亥姆霍兹面(IHP)和外亥姆霍兹面(OHP)。通过添加催化中心,S正极表面的EDL组分被重构。由于催化剂对Mg2+的去溶剂化和吸附的催化作用,Mg(solvents)x2+结构将被吸引向正极/电解质界面移动。当Mg(solvents)x2+结构与S正极表面接触时,由于催化剂对未来转化反应的催化作用,Mg2+经历快速去溶剂化。与IHP中溶剂分子显著增加的空白样品相比,催化剂将大量游离Mg2+和多硫化物限制在三相界面,进一步加速了后续的硫/硫化物转化。
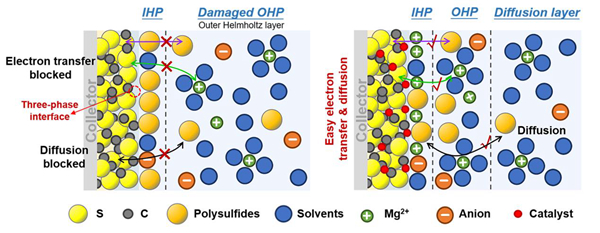
图3. 无/有催化剂的情况下正极/电解质界面处的EDL结构的示意图
与Li-S电池类似,Mg-S电池也遇到电化学动力学差和可溶性多硫化物穿梭的问题,导致低比容量、低循环寿命和低库仑效率。为了获得实用的高性能Mg-S电池,了解反应动力学显得非常重要。在硫/硫化物氧化还原反应的充放电过程中,从固态硫到液态多硫化物再到固态硫化镁的相变明显,反应势垒较大。同时,相变也导致缓慢的电化学动力学和可溶性多硫化物的积累。因此,有必要对可溶性多硫化物进行定点吸附,进而促进多硫化物的转化,“吸附+转化”的催化策略可以有效抑制多硫化物的穿梭,降低转化反应势垒,增强反应动力学。
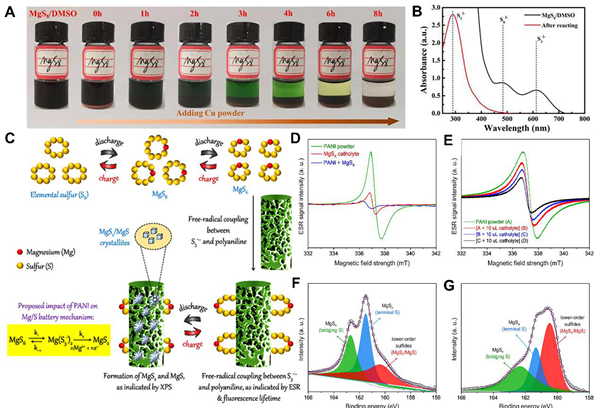
图4. 催化硫/多硫化物转化的策略
以上论文成果的通讯作者为王健博士和蔺洪振研究员,第一作者为中国科学院苏州纳米所博士生关青华,论文以Prospect of Cascade Catalysis in Magnesium-Sulfur Batteries from Desolvation to Conversion Reactions为题,发表在Advanced Science期刊中。以上联合工作受到国家重点研发计划、国家自然科学基金、中国博士后科学基金、江苏省自然科学基金、江苏省重点研发计划、教育部重点实验室开放资金及德国洪堡基金等项目支持,受到了中国科学院苏州纳米所Nano-X的表征技术支持。
 扫一扫关注我们
扫一扫关注我们